|
|
|
Home | Pregnancy Timeline | News Alerts |News Archive Jun 4, 2015
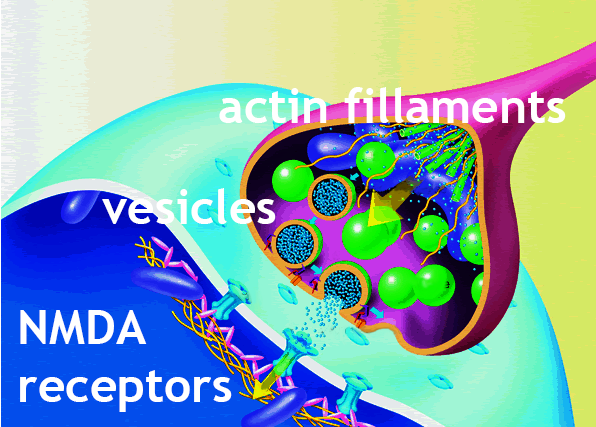
A Shank3 gene deficiency disrupts NMDA receptors — not allowing them
to open and receive impulses from [orange] actin filaments during synapse.
The Shank3 mutation also disrupts actin filament
regulators, not allowing for
the normal assembly and disassembly of filaments.
|
|
|
|
|
|
Genetic mutation link to autism might be undone
Original article by Ellen Goldbaum, University at Buffalo
Scientists at the University at Buffalo have identified the mechanisms behind a genetic mutation that produces certain autistic behaviors in mice, as well as therapeutic strategies that restored normal behaviors in the mice.
Research led by Zhen Yan PhD, professor in the Department of Physiology and Biophysics at the University of Buffalo (UB) School of Medicine and Biomedical Sciences, describes cellular and molecular processes behind some autistic behaviors. It also suggests potential biomarkers and pharmaceutical targets for treatment.
The work was published May 28 in Cell Reports.
“Our results suggest a promising therapeutic strategy for treating autism.”
Zhen Yan PhD, professor, Department of Physiology and Biophysics, UB School of Medicine and Biomedical Sciences.
The loss of a gene called Shank3, is an important risk factor for autism spectrum disorders (ASDs). Previous studies have shown that approximately 84 percent of people with a Shank3 deletion, a mutation leading to loss-of-function in Shank3, have an Autism Spectrum Disorder (ASD). But why was unknown.
Yan and his team traced the Shank3 loss and found that it disrupts communication between neurons, particularly those neurons leading to social behaviors — such as mutual grooming — in mice.
A Shank3 deficient mice exhibit “drastically reduced” interest in social stimuli from other mice, causing a “severe social deficit.” These mice spent significantly more time repetitively grooming them selves, than normal mice.
But, their most important finding was the ability to reverse these disruptions and restore normal mouse behaviors. When UB researchers found that Shank3 is key to how neurons communicate, because it affects the activation of a receptor called n-methyl-D-aspartate (NMDA) critical to learning and memory, they were then able to supplement the missing amino acids.
N-Methyl-D-aspartic acid or N-Methyl-D-aspartate (NMDA) is an amino acid that behaves as an agonist on the NMDA receptor, opening it up (mimicking the action of glutamate). Unlike glutamate though, NMDA only binds to and regulates the NMDA receptor with no effect on other receptors. These receptors are particularly noticable when they become overactive during withdrawal from alcohol — causing agitation and sometimes, epileptic seizures.
“This research is the first to show that in animals, abnormal actin regulation causes autism-like behaviors. Actin filaments are very dynamic structures constantly being assembled and disassembled, processes controlled by numerous regulators,” Yan explained. When the equilibrium between actin filament assembly and disassemby is upset, signal transmissions fail.
“With Shank3 deficiency, we found the expression (or activity) of some actin regulators — such as cofilin — is altered. This upsets the equilibrium of the actin filament assembly, which in turn disrupts normal delivery and maintenance of NMDA and other critical receptors.”
This results in a significant negative effect on the plasticity of the synapse, leading to some autistic behaviors.
Zhen Yan PhD
As cofilin or other regulators were returned to normal, actin was restored, allowing for the normal trafficking and functioning of the NMDA receptors.
“Once actin filaments and NMDA receptors returned to normal, we observed a robust and long-lasting rescue of the social interaction deficits and repetitive behavior in the Shank3-deficient mice. Our results suggest a promising therapeutic strategy for treating autism.”
Zhen Yan PhD
The researchers are seeking funding to continue their work developing potential biomarkers and treatments for autism.
Abstract
Highlights
•Shank3 deficiency induces ASD-like behavioral deficits and NMDAR hypofunction in PFC
•Shank3 deficiency leads to reduced synaptic F-actin and altered actin regulators in PFC
•Inhibiting cofilin rescues behavioral and synaptic deficits in Shank3-deficient mice
•Manipulating cortical Rac1 or PAK controls the manifestation of ASD-like phenotypes
Summary
Haploinsufficiency of the Shank3 gene, which encodes a scaffolding protein at glutamatergic synapses, is a highly prevalent and penetrant risk factor for autism. Using combined behavioral, electrophysiological, biochemical, imaging, and molecular approaches, we find that Shank3-deficient mice exhibit autism-like social deficits and repetitive behaviors, as well as the significantly diminished NMDA receptor (NMDAR) synaptic function and synaptic distribution in prefrontal cortex. Concomitantly, Shank3-deficient mice have a marked loss of cortical actin filaments, which is associated with the reduced Rac1/PAK activity and increased activity of cofilin, the major actin depolymerizing factor. The social deficits and NMDAR hypofunction are rescued by inhibiting cofilin or activating Rac1 in Shank3-deficient mice and are induced by inhibiting PAK or Rac1 in wild-type mice. These results indicate that the aberrant regulation of synaptic actin filaments and loss of synaptic NMDARs contribute to the manifestation of autism-like phenotypes. Thus, targeting actin regulators provides a strategy for autism treatment.
This is an open access article under the CC BY-NC-ND license
Yan’s co-authors are Lara Duffney, PhD, former doctoral student; Ping Zhong, PhD, research assistant professor; Jing Wei, PhD, research assistant professor, Emmanuel Matas, PhD, postdoctoral associate; Jia Cheng, PhD, former doctoral student; Luye Qin, research scientist and Kaijie Ma, research assistant, all in the UB Department of Physiology and Biophysics; also, David Dietz, PhD, assistant professor in the UB Department of Pharmacology and Toxicology; and Yuji Kajiwara and Joseph D. Buxbaum of the Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai.
- See more at: http://www.buffalo.edu/news/releases/2015/05/045.html#
sthash.0NQbWnqY.dpuf
Return to top of page
|


