|
|
Correcting gene error restores brain function
Gene duplications are a common cause of intellectual disabilities. Correcting gene duplication by eliminating excess duplicated genes in critical brain regions, may have potential for treatment of intellectual disorders.
In a new study that appears online in the journal Nature, Dr. Huda Zoghbi, professor of molecular and human genetics at Baylor College of Medicine and director of the Jan and Dan Duncan Neurological Research Institute at Texas Children's Hospital, with her team reveals there is a potential way to treat gene brain duplication disorders.
Me0CP2 (methyl-CpG-binding protein) is a maestro at controlling the expression of thousands of brain genes. But its own level within the brain must also be controlled. Too little of the protein results in Rett syndrome, a neurological disorder characterized by decreased comprehension, inability to perform some motor functions, and autism-like behavior.
More than 10 years ago, Zoghbi, a Howard Hughes Medical Institute investigator, discovered MeCP2 is a "Goldilocks" protein - too little causes Rett syndrome, but too much can cause a different neurological problem.
The mouse models Zoghbi's lab generated with an extra copy of the MeCP2 gene developed a progressive neurological disorder. She suspected there must be others with comparable neurological problems due to duplication of MECP2 and found this to be the case.
Boys with MECP2 duplication syndrome suffer from poor muscle tone and motor function, cognitive disability, epilepsy, autistic behaviors, respiratory infections, and premature death.
Once considered rare, it appears the disorder is more common than previously thought, said Dr. Yehezkel Sztainberg, a post-doctoral fellow in Zoghbi's laboratory and first author of the paper.
Using both state-of-the-art genetic techniques and a small molecule that can target specific genetic material, Zoghbi, Sztainberg and team have shown it is possible to reverse the terrible effects of a duplicated gene.
In a series of experiments, they have showed that mice engineered to have an extra copy of MECP2 develop all the symptoms of the human disorder. However, when such mice became fully-symptomatic, deleting the extra MECP2 gene normalized the levels of MeCP2 and reversed many of the phenotypes "providing the proof of principle that the disorder is reversible in adult animals," Sztainberg explained.
However, while deleting the gene proved the concept that the neurological dysfunction in mice can be reversed, it is not yet a feasible treatment for human patients.
To develop a translational tool, Zoghbi and her colleagues turned to AntiSense Oligonucleotides (ASOs). They found normalizing levels of MeCP2 with ASOs largely reversed behavior, molecular, and other deficits afflicting the mice. After treating young adults, researchers used ASOs on older mice who by that time were having many seizures. With the ASO treatment, their seizures stopped. "That was very encouraging," Zoghbi added, as it meant brains of the mice were not permanently damaged and could recover.
ASOs are currently being tested in individuals with spinal muscular atrophy, a devastating genetic disorder, providing hope the MECP2 duplications approach can be used in humans in the future.
ASOs take advantage of the most basic biology of genetics — the base adenine (A) always seeks out and pairs with the base thymine (T) in DNA, or uracil (U) in RNA and cytosine (C) pairs with guanine (G).
When antisense oligonucleotides (ASOs) — small chemically modified nucleic acids — pair up with messenger RNA transcribed from a gene, they silence the gene.
This corrects deficits caused by excess MeCP2. In other words, the antisense base pairs seek out their natural partners and silence the extra gene.
The antisense treatment was developed in collaboration with Isis Pharmaceuticals and targets the human copy of the gene.
This approach has potential for treating other disorders in which there is duplication of genetic material by targeting genes in critical regions, adds Sztainberg. Among these are autism and intellectual disabilities, Charcot-Marie-Tooth disease, Potocki-Lupski syndrome, as well as Down syndrome, the most common of these disorders. Treating Down syndrome, in which an entire chromosome is duplicated, would require determining the critical gene(s).
Sztainberg: "Now it's translational," meaning now their basic research findings can be applied to enhance human health and well-being.
"More work must be done in the animal model before we consider clinical trials. The additional experiments are to determine how to titrate and monitor the dose of ASOs to safely normalize MeCP2 levels without lowering them below the normal range to avoid causing Rett-like symptoms."
Huda Zoghbi MD, Professor, Departments of Pediatrics, Molecular and Human Genetics, Neurology, and Neuroscience, Baylor College of Medicine; Investigator, Howard Hughes Medical Institute; Member, Institute of Medicine and National Academy of Sciences
"Down syndrome is the prototypical gene dosage disorder," said Zoghbi.
While Downs results from a duplication of an entire chromosome, using antisense oligonucleotides to knock out some of its critical genes affecting learning, memory as well as dementia, could prove important.
Abstract
Copy number variations have been frequently associated with developmental delay, intellectual disability and autism spectrum disorders1. MECP2 duplication syndrome is one of the most common genomic rearrangements in males2 and is characterized by autism, intellectual disability, motor dysfunction, anxiety, epilepsy, recurrent respiratory tract infections and early death3, 4, 5. The broad range of deficits caused by methyl-CpG-binding protein 2 (MeCP2) overexpression poses a daunting challenge to traditional biochemical-pathway-based therapeutic approaches. Accordingly, we sought strategies that directly target MeCP2 and are amenable to translation into clinical therapy. The first question that we addressed was whether the neurological dysfunction is reversible after symptoms set in. Reversal of phenotypes in adult symptomatic mice has been demonstrated in some models of monogenic loss-of-function neurological disorders6, 7, 8, including loss of MeCP2 in Rett syndrome9, indicating that, at least in some cases, the neuroanatomy may remain sufficiently intact so that correction of the molecular dysfunction underlying these disorders can restore healthy physiology. Given the absence of neurodegeneration in MECP2 duplication syndrome, we propose that restoration of normal MeCP2 levels in MECP2 duplication adult mice would rescue their phenotype. By generating and characterizing a conditional Mecp2-overexpressing mouse model, here we show that correction of MeCP2 levels largely reverses the behavioural, molecular and electrophysiological deficits. We also reduced MeCP2 using an antisense oligonucleotide strategy, which has greater translational potential. Antisense oligonucleotides are small, modified nucleic acids that can selectively hybridize with messenger RNA transcribed from a target gene and silence it10, 11, and have been successfully used to correct deficits in different mouse models12, 13, 14, 15, 16, 17, 18. We find that antisense oligonucleotide treatment induces a broad phenotypic rescue in adult symptomatic transgenic MECP2 duplication mice (MECP2-TG)19, 20, and corrected MECP2 levels in lymphoblastoid cells from MECP2 duplication patients in a dose-dependent manner.
Zoghbi is also a professor of pediatrics and neuroscience. Others who took part in this work include Hong-mei Chen, John W. Swann, Shuang Hao, Bin Tang, Zhenyu Wu, Jianrong Tang, Ying-Wooi Wan, and Zhandong Liu all of Baylor and the Neurological Research Institute, and Frank Rigo of Isis Pharmaceuticals in Carlsbad, California.
Competing financial interests
Frank Rigo is an employee of Isis Pharamaceuticals.
Funding for this work came from National Institutes of Health (Grant 5RO1NS057819 and 5P30HD024064), the Rett Syndrome Research Trust (401 Project), the Carl. C. Anderson Sr. and Marie Jo Anderson Charitable Foundation, the Howard Hughes Medical Institute, the National Science Foundation (Grant DMS-1263932) and the Baylor Intellectual and Developmental Disabilities Research Center (Grant 1U54HD083092).
Related articles: Chromosome errors may explain brain birth defects
Return to top of page
|
|
|
Nov 30, 2015 Fetal Timeline Maternal Timeline News News Archive
BLUE outline indicates location of hipocampus in a mouse brain
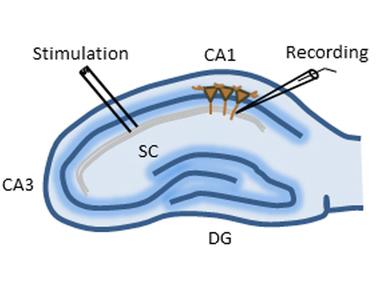
A diagram of a mouse hippocampus being manipulatied through the application
of high-frequency stimulation to nerve axons. An Excitatory PostSynaptic Potentials
(fEPSPs) — or more simply, an electrical current — was then recorded in the CA1 synapses.
Image Credit:
Nature
|
|
| |
|


