|
|||||||||||||||
|

CLICK ON weeks 0 - 40 and follow along every 2 weeks of fetal development
|
||||||||||||||||||||||||||||
Genes can cause neurological diseases Currently, SCA has no cure or treatment and genetic mutations responsible for about 30 percent of cases are still unidentified. However, two different families with inherited SCA looking for treatment and unable to be helped, had gene samples sent to Hiroshima University to begin the process of identifying their new mutation. After sequencing the genes of family members with SCA, a research team — led by Professor Hideshi Kawakami MD PhD, from the Department of Epidemiology at Hiroshima University — began statistical analysis comparing family members' DNA to unrelated people without SCA. The analysis identified which gene variation SCA family members shared that is not found in healthy people.
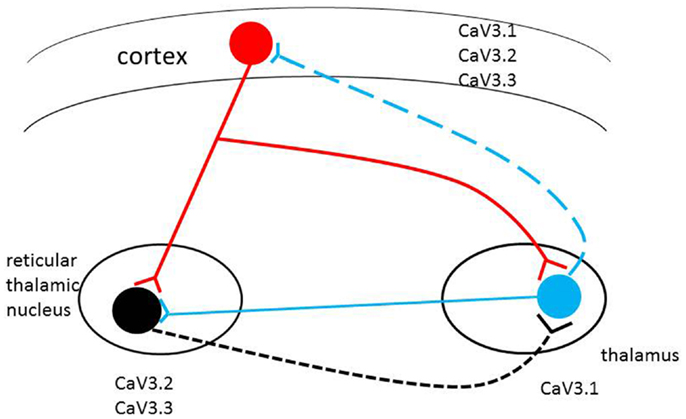
Researchers performed more experiments in culture to examine how the mutated Cav3.1 channel behaves and found this mutation makes calcium channels open at a lower threshold than in healthy cells. "In the future, a drug modifying this channel may cure patients," suggests Prof. Kawakami. Skin cells from one patient were used in generate pluripotent stem cells, allowing for a patient's neurons to be grown in the laboratory. These newly grown neurons showed no obvious physical deformities, which might fit with the normal progression of SCA. Depending on which SCA mutation a patient has, some patients may not experience symptoms until they are middle-aged. Prof. Kawakami: "We might need some age-related factors to reproduce life-like cell behavior," so research will continue using these lab dish neurons to study Cav3.1 under more life-like conditions and in greater detail in the future. T-type calcium channels are low-voltage. They open during membrane depolarization, mediating the amount of calcium flowing into cells. After being given the depolarizing signal, the amount of calcium flowing into a cell induces numerous physiological responses.
Calcium channels in the 1970s became distinguished in cells of the central nervous system as either Transient Opening calcium channels (T-type calcium channels) or the more well-known Long-Lasting calcium channels (L-type calcium channels).
Through this pore, T-type calcium channels allow for the continuous rhythmic bursts of calcium opening valves in the heart, as well as for relaying rapid signal transmissions from the thalamus to numerous other locations throughout our body. And, as there is pharmacological evidence supporting these channels as key players in epilepsy, diabetes, and several forms of cancer, research continuously looks to create drugs to regulate their influence when irregular or missing. Abstract Results (Open Article Online) Conclusions |
Mar 17, 2016 Fetal Timeline Maternal Timeline News News Archive
|
||||||||||||||||||||||||||||